April 2022
Abstract
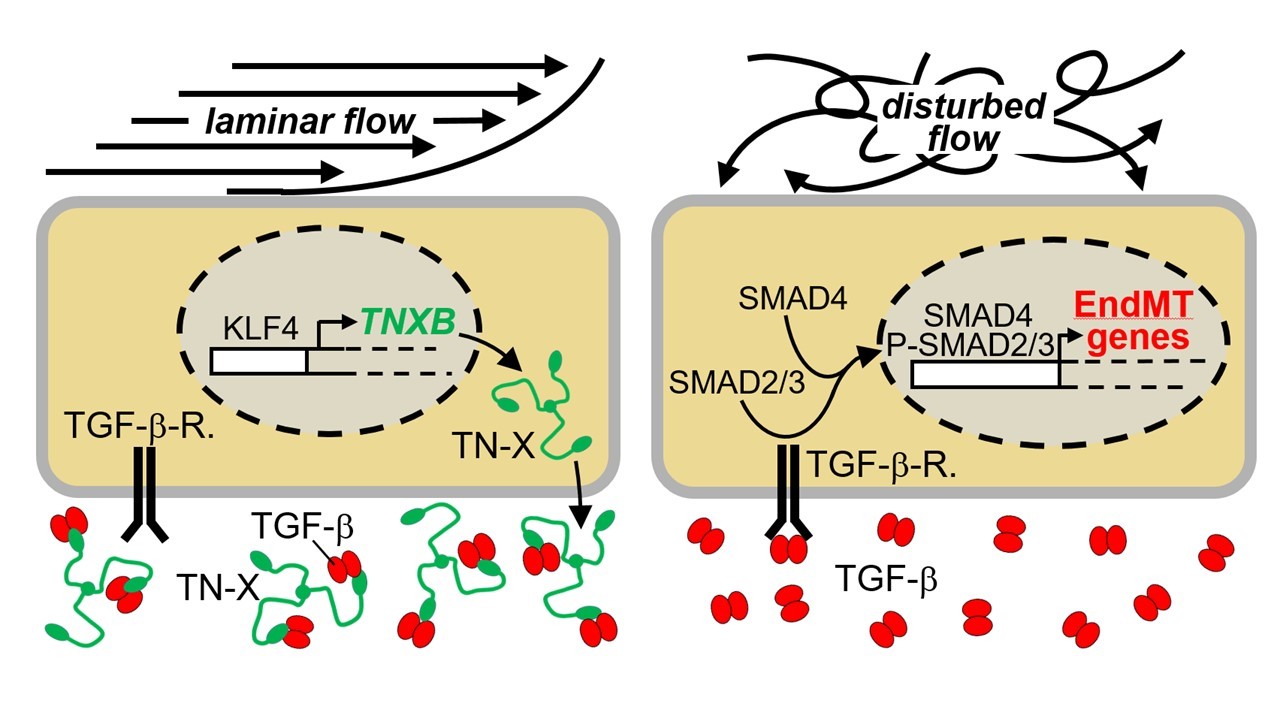
Endothelial-to-mesenchymal transition (EndMT) has been identified as a critical driver of vascular inflammation and atherosclerosis, and transforming growth factor β (TGF-β) is a key mediator of EndMT. Both EndMT and atherosclerosis are promoted by disturbed flow, whereas unidirectional laminar flow limits EndMT and is atheroprotective. How EndMT and endothelial TGF-β signaling are regulated by different flow patterns is, however, still poorly understood.
In this study, we show that unidirectional laminar flow but not disturbed flow strongly increased the endothelial expression of the extracellular matrix protein tenascin-X (TN-X) in vitro as well as in human and mouse aortae and that this effect was mediated by Krüppel-like factor 4 (KLF4). Mice with endothelium-specific loss of TN-X (EC-Tnxb-KO) showed increased endothelial TGF-β signaling as well as increased endothelial expression of EndMT and inflammatory marker genes. In addition, EC-Tnxb-KO mice also showed increased vascular remodeling after partial carotid artery ligation as well as accelerated atherosclerotic lesion development. Treatment of EC-Tnxb-KO mice with an anti-TGF-b antibody or additional endothelial loss of TGF-b receptors 1 and 2 normalized endothelial TGF-b signaling and prevented EndMT. In in vitro studies, we found that TN-X through its fibrinogen-like (FBG) domain directly interacts with TGF-β and thereby interferes with its binding to the TGF-β receptor. In summary, we provide evidence that TN-X is a central mediator of flow-induced inhibition of EndMT, endothelial inflammation and atherogenesis, which functions by binding to and by blocking the activity of TGF-β.
Legend to graphical abstract:
Schematic representation showing the flow pattern-dependent function of TN-X in protecting endothelial cells from TGF-β-induced EndMT. Unidirectional laminar flow induces expression of endothelial TNXB. The homotrimeric TN-X binds via its C-terminal FBG domain to TGF-b and thereby prevents TGF-b from binding to its receptor on endothelial cells.